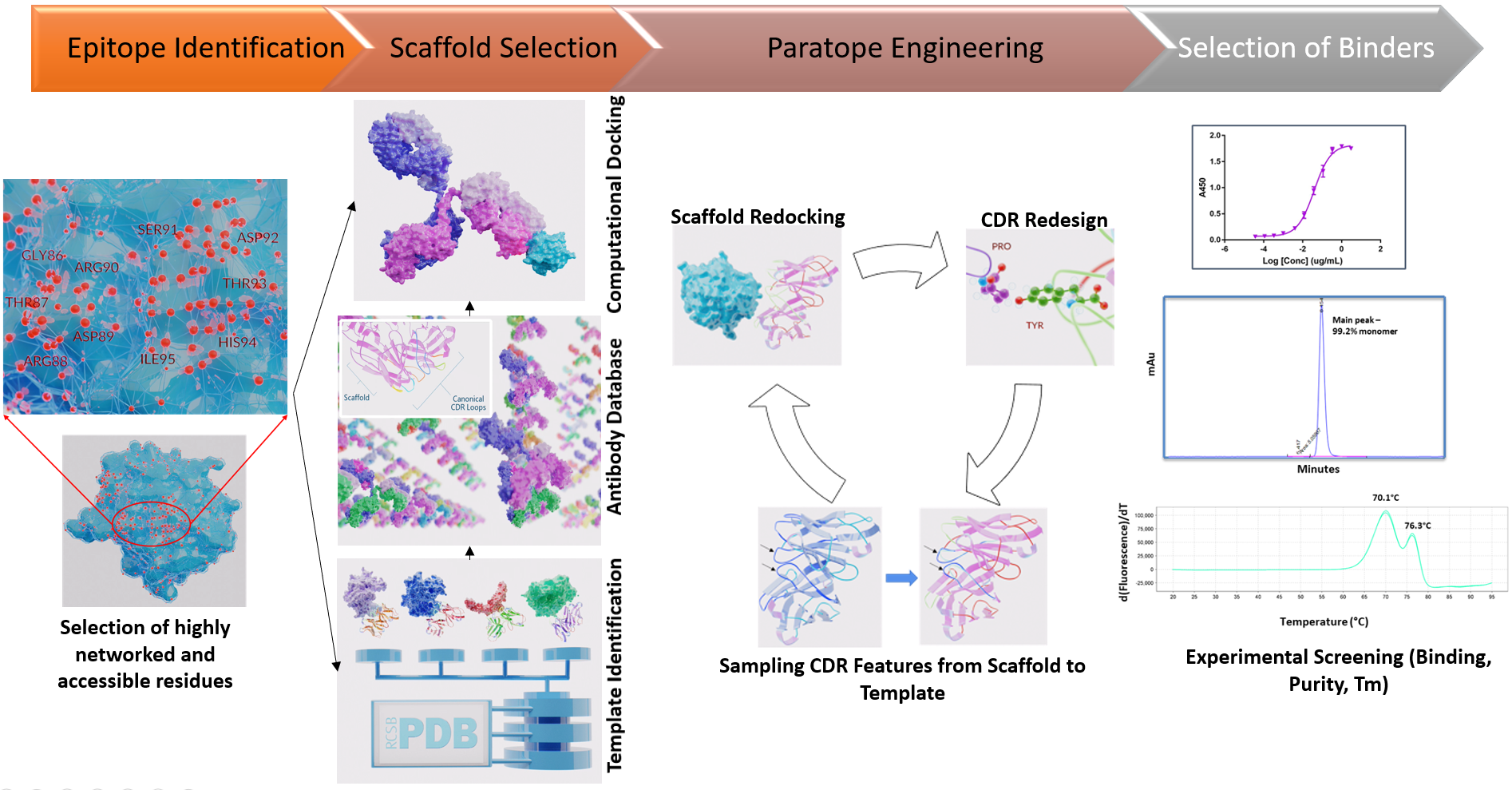
Brief Description of the Antibody Engineering Platform:
The 4-part strategy of antibody engineering (referred here as 3DMAbDesign) involves: 1) Target Epitope Identification, 2) Scaffold Selection, 3) Paratope Engineering and 4) Selection of Binders. Parts 2) and 3) involve selection, modification, and assembly of antibody parts (framework region or FWRs and all 6 complimentarity determining regions or CDRs of heavy and light chains) by searching millions of antibody sequences and structures of antibodies and antibody-antigen complexes from publicly accessible data repositories. The scaffold selection step in part 2) involves identifying the ideal starting scaffold of heavy and light chain (which can each come from the same antibody or two different antibodies) for optimal engagement with the target epitope. The paratope engineering step in part 3) comprises of iterative cycles of selecting appropriate CDR features including loop lengths and residue modifications to optimize the AIF (also referred to as Interface Fitness Score or IFS in the animation), which measures the inter-residue contact network between epitope and paratope in the context of the starting scaffold. These iterative cycles result in antibody designs that are subsequently experimentally tested for antigen binding and other properties including developability in part 4) of the strategy.If none of the designs meet the experimental screening in part 4), then a new starting scaffold from the list of top-ranking scaffolds is chosen and the paratope engineering is performed in the context of this new starting scaffold. Importantly, this process proceeds in a parallel fashion with early experimental readouts providing feedback to the mAb engineering to rapidly identify one or more lead candidates
See an animation of the 3DMAbDesign platform in action (click here) to get an overview first.
The following describes key steps involved in our 4-part strategy
Part 1: Target Epitope Identification
Input: X-ray crystal structure or cryo-EM structure of a target protein OR a primary amino acid sequence of the target, which can be modeled into a 3D structure using a homology modeling approach
- Compute the network score and solvent accessible surface area (SASA) for the individual residues of the input protein structure. If the structure contains multiple identical chains arranged in a symmetric manner (e.g. 532), treat the chains as different units according their proximity to the symmetry axes (e.g. 2-fold chain, 3-fold chain, 5-fold chain) and catalog the network scores and SASA values separately. SASA calculation: DSSP (https://swift.cmbi.umcn.nl/gv/dssp/), POLYVIEW-2D (https://polyview.cchmc.org/).
- Select residues that are highly networked, solvent-accessible, and that overlap with a known functional region.
Part 2: Scaffold Selection
Input: Target epitope surface in 3-D coordinates which has sequence and structural information
- Searching antibody structural databases such as PDB, electron microscopy databank for template antibodies and their associated features (CDRs and/or FWRs) that can potentially bind to the target epitope surface
- Search criteria: identify template antibodies and associated features that bind to epitope motifs that are homologous to regions of the target epitope surface. Homology criteria include a minimum threshold sequence similarity between the query and target epitopes and a maximum RMSD upon structural superimposition of the query epitope to the target epitope.
- Databases: PDB, SAbDAB .
- Epitope Search and comparison PDBePISA, PyMOL, BioPython, PyRosetta.
- Results: List of template antibodies satisfying these criteria <Template mAbs>.
- Use <Template mAbs> to search larger mAb sequence databases to identify list of antibody scaffolds that will give the starting point for engineering the new antibody
- Search criteria: Antibodies should contain CDR loops belonging to similar canonical classes as the CDR loops in the <Template mAbs>, exhibit shape complementarity against target epitope (Steps 3-4) and should meet an initial developability criteria such as abundance of predicted T-cell epitopes and net charge asymmetry (product of net VH and VL charges, PMID: 25512516). On a case-by-case basis, depending on the depth of the template or target structural data, users adjust the stringency of the canonical constraints. If no canonical constraints are applied, then the antibody scaffold selection will be guided only by the developability criterion.
- DBs and structural annotation resources: PyIgClassify, Abysis, Martin Group, NCBI.
- T-cell epitope prediction tools – NETMHCIIpan 4.0
- Results: List of several scaffold mAbs that meet the above criteria <Scaffold mAbs>.
- Generate homology based structural models for those mAbs in <Scaffold mAbs> that don’t have X-ray or cryo-EM structure
- Homology modelling tools: Abodybuilder, Rosetta Antibody, SWISS-MODEL.
- Computational docking of <Scaffold mAbs> to target epitope.
- Epitope-paratope contacts are first defined based on metrics such as AIF (also referred to as interface fitness score or IFS in the animation) and these contacts are subsequently uased as constraints to guide the docking process.
- Computational docking software includes: ZDOCK; PATCHDOCK; and RosettaAntibody.
- Poses are ranked using previously validated metrics including shape complementarity, buried SASA and paratope location in accordance with knowledge of existing antibody-antigen complexes of Abs engaged with regions partially overlapping the target epitope.
- Interfaces are analyzed for buried SASA, bonds, and free energy using PDBePISA and shape complementarity is computed by the docking software or other software such as PyRosetta .
- Pick top ranking scaffold as starting scaffold <Start_scaffold>.
Part 3: Paratope Engineering
Input: Initial model of <Start_scaffold> with target epitope
- Assembling the optimal CDR features from <Template mAbs> into the <Start_scaffold> (referred to as grafting in the animation) is carried out in the following stepsand introduce mutations in both CDRs and FWR to optimize the epitope-paratope contacts based on initial model
- Feature assembly: Select features from <Template mAbs> and introduce mutations one at at time if needed starting from most exposed to most buried residues in the selected CDRs and also introduce mutations in FWR if needed (to refine the interfaces between heavy and light chains and framework and CDRs).
- Scoring assembled features: Amino Acid Substitutions are guided by AIF or IFS score, binding energy, visual inspection, EPC network and CDR-FWR connectivity network.
- Tools to model assembled features: CDR sampling and amino acid substitutions can be introduced by homology modelling (SWISS-MODEL, ABodybuilder), PyMOL or through RosettaAntibody.
- Modified VH/VL redocked with target epitope to generate the new input starting model, which is selected using multivariate logistic regression (MLR) method.
- Repeat step 1 and 2 iteratively with each iteration representing a design candidate until additional modifications deteriorate the scoring metrics.
- Submit all the designs which pass set threshold scores for experimental screening as described below.
Part 4: Selection of Binders
- Protein parameter screening: Recombinantly express full-length antibody for each candidate and screen for the following protein parameters – expression yield, purity, solubility and aggregation, and if none of the candidates pass threshold levels, repeat scaffold selection and paratope engineering steps with alternate scaffolds among the top candidates for shape complementarity.
- Binding affinity screening: Constructs that pass the key threshold metrics for protein parameters are next assessed for their binding affinity to the antigen comprising the target epitope. If all constructs fail, then repeat caffold selection and paratope engineering steps with alternate scaffolds that are among the top candidates for shape complementarity.
The iterative cycle of scaffold selection, paratope engineering and experimental screening converges to a set of one or more lead antibody constructs with all the desired properties including developability parameters.
For a complete list of available tools for use in the above methods see: tools