Building on our previous analyses, we have identified key amino acid positions and its contact network that influence GAG-binding specificity of proteins and enzymes [9, 10]. For example, based on network analyses of Heparinase II –substrate co-crystal structures [11], we identified specific positions in the active site that were involved in contacts with the GAG substrate and whose residue contacts network was such that they could be mutated to alter the substrate specificity of this enzyme. Among these positions, 405 and 436 critically govern interactions with the 3-OH group of the glucosamine residue in the cleavable linkage; hence mutations at these positions would enable the Hep II mutant to completely depolymerize heparins and LMWHs.
These computed properties along with information on GAG-related proteins are captured in the informatics platform and serve as useful parameters for generating variants of these proteins with desired GAG-binding specificities. In the case of GAG-binding proteins, we analyzed network properties of the best-studied members of the FGF family. While it is generally known that the residues involved in heparin binding are highly variable between different FGFs (accounting for the structural specificities to diverse heparan sulfate motifs), we identified key anchor residues in the heparin-binding site whose network properties were the same across members of the FGF family (Figure 6). Identification of this feature pointed to a common mode of HSGAG recognition by all FGFs.
Going beyond our established studies on heparinases and FGF, we also investigated the role of HSGAGs as (co-) receptors for pathogens. One such area of immediate public health concern, was to investigate the role of HSGAGs in human infection by dengue viruses (DV) [12]. While it was generally known that envelope protein (gpE) on DV interacts with HSGAGs on the surface of human host cells, studying this interaction in the context of natural DV has been complicated, given that studies have shown that DVs or recombinant gpEs produced in cells readily acquire mutations that affect HSGAG binding. We addressed this question by developing a system using recombinantly generated sub-viral particles (RSVs) in collaboration with the Chulabhorn Research Institute, Bangkok Thailand. RSVs afford the ability to introduce defined mutant forms of gpE without such mutations being altered by cell culture passage and also allowing presentation of gpEs on surface of the particle in a fashion similar to authentic DV. By performing biochemical binding of RSV-HSGAGs and in vitro competitive inhibition of DV infection of cells with HSGAGs, we were able to provide strong evidence to support the role of HS as co-receptor in infection with minimally passaged clinical DV isolates.
We also extended our approach to investigate other glycan-protein systems going beyond GAGs. In collaboration with Mark Bulmer (now at Towson University), we came across a unique pattern recognition receptor in termites (tGNBP-2), which was known to play a role in their antimicrobial defense, but its mechanism of action was not fully known. Given the lack of crystal structure of tGNBP-2, we used our PCIN approach to construct a structural model of tGNBP-2. Using this model, we showed that this protein had distinct binding sites for glucan polysaccharides and LPS. The glucan-binding site of tGNBP-2 also had characteristic features shared by glucanases, enzymes that depolymerize glucans. Based on this model, we determined a sugar-based molecule (D-δ-gluconolactone or GDL) that effectively inhibited glucanase activity of tGNBP-2 and lead to dramatically reduced termite survival as measured both in vitro and in vivo by weakening its defense mechanisms (see [13] for details).
Finally, in collaboration with Momenta Pharmaceuticals, we demonstrated structure-function relationship of a novel CHO α-galactosyl transferase that had important implications on biopharmaceutical manufacturing [14]). Significant biomanufacturing occurs in CHO cells, and this source has long been assumed to be incapable of placing immunogenic 1-3 alpha galactose on glycosylated proteins. In this study, we showed that CHO cells do indeed have α-galactosyl transferase activity, identified the putative gene, and through our structural approach definitely identified the fold, active site, and putative mechanism of action.
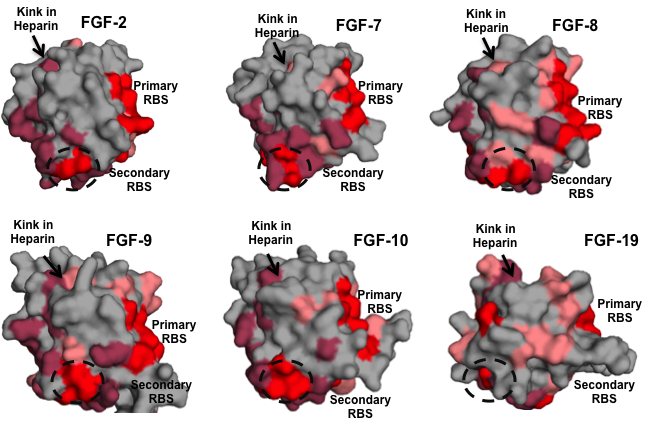
Figure 6. Mapping key sites on FGFs
Using the network approach, we map key sites on the different FGF.