Our focus has also been to develop an informatics platform to enable the integration of analytical methods and facilitate the characterization of complex glycan mixtures. Towards this goal, we have created and continue to populate a database of GAG sequences and have developed web-based interfaces to query this database using molecular weight, composition and substructure of the glycan. Through our involvement in international collaborative efforts on glyco-informatics (Consortium for Functional Glycomics), we have collectively defined and implemented a XML format (Glyde-II) to represent and exchange glycan structural information and developed web-services to accept XML-based queries using Glyde-II format for molecular weight, composition, and substructure searches to the glycan structure. We anticipate that developing these web-services will permit data exchange and interoperability with other glycomics databases, a critical step towards developing a sustainable, general informatics platform for glycans. To address the function ‘attribute’ in the structure-function relationship of complex glycan mixtures such as GAGs, we found that it was necessary to understand the molecular and structural basis, which leads to specificity in glycan-protein interactions. The proteins that interact with glycans are of two kinds: (1) glycan-binding proteins that mediate recognition and downstream events and (2) glycan modifying enzymes that serve as tools to characterize glycan structure. We have expanded this informatics platform and developed a novel framework to model glycan-protein interactions and get to the heart of glycan-protein structure-function relationships in important areas with practical impact.
Describing protein structure and fold using amino acid interaction networks – Novel Applications
Obtaining a structural model of protein-glycan complexes is a key step towards broadly understanding the basis of specificity. While X-ray co-crystal structures are available for many protein-glycan complexes including protein-GAG oligosaccharides, there are many instances where the crystal structure of protein is not available. In these situations, it is necessary to model the protein structure. Homology modeling of three-dimensional protein structures is a valuable tool when assigning the most likely structure of a given protein based on its sequence identity with another protein whose X-ray crystal structure has already been solved. However, there remains a ‘twilight zone’ where for any two proteins that share < 25% sequence identity; there are significant inaccuracies in homology model-based structure assignment. The twilight zone issue is especially prominent for glycan-binding proteins (GBPs) due to their large sequence divergence. Through analysis of the atomic-level interactions between amino acids within the protein core (not exposed to solvent) of common structural folds adopted by GBPs, we developed a new scoring system to evaluate the protein core interaction network (PCIN) for a given structural fold [5]. The score was computed by normalizing the number of contacts between two residues within the protein core. We constructed a database of such scores for all the protein fold families using structural alignment of crystal structures from PDB and sequence alignment data from DALI database. Using this scoring system we were able to generate more accurate models of GBPs, which shared very low sequence identity to any known protein structure (Figure 4).
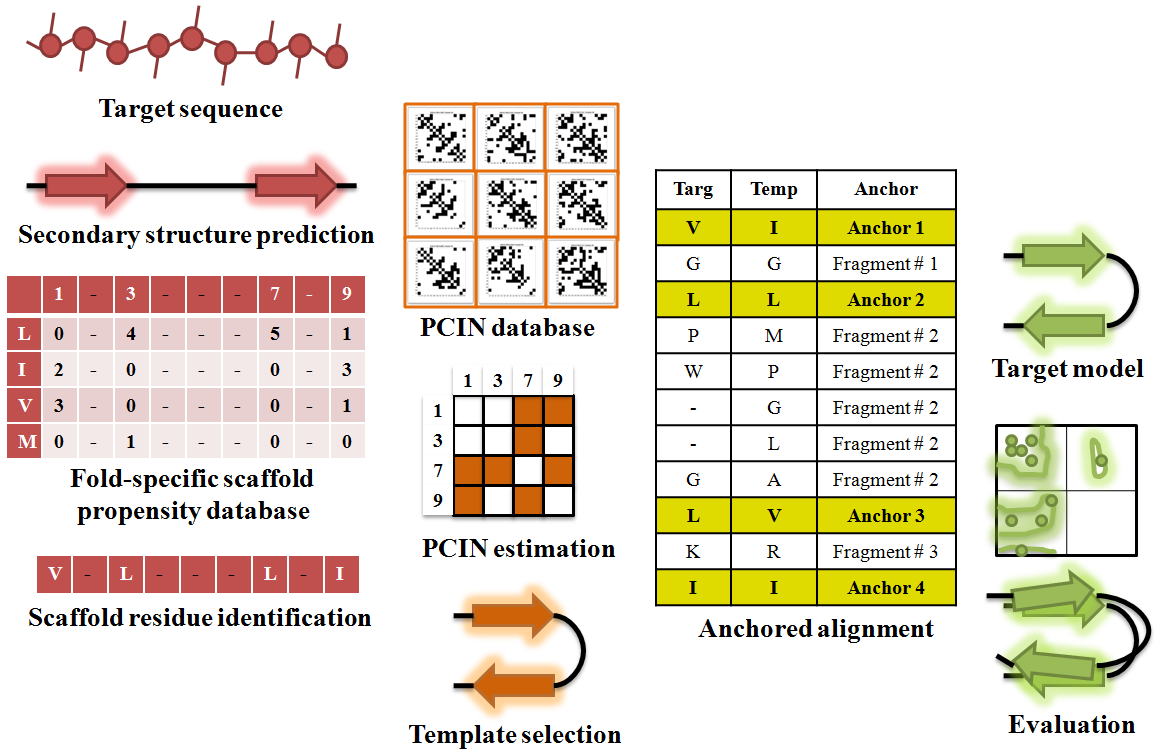
Figure 4. Modeling 3-D structure of GBP using PCIN
Predicted secondary structure from sequence is used to filter the structural template database to identify appropriate templates, which are further filtered, based on PCIN to select appropriate template for most accurate model.
We further developed the framework of analyzing atomic interactions within the protein core towards systematically capturing and quantifying inter-residue interactions (IRI) using concepts from network theory (Figure 5). IRI — including hydrogen bonds, disulfide bonds, pi-bonds, polar interactions, salt bridges, and van der Waals interactions — are computed between all pairs of amino acid residues within a protein structure. These interactions are used to generate a network map known as a residue contacts network of residues that had significant contacts with other residues in their environment (see [6] for details). The degree of networking (i.e. the extent of contacts of a residue with other residues in its environment) is captured using a contacts score that, when normalized, varies from 0 (minimum) to 1 (maximum). The residue contacts map thus is a novel way to visualize protein structure [7], providing a complementary view to physics-based approaches, and which has a number of important elements. First, the extent of networking (captured by the network score) of an amino acid is related to the structural constraints imposed on its ability to change (or mutate) either naturally or engineered to achieve a specific functional outcome. Second, the 2-D contact map goes beyond looking at contacts made by individual ‘hallmark’ amino acids with glycans to provide a broader picture of features of glycan-binding site that include the connectivity between amino acids, which governs how variation of amino acids at a given position in the structural space is incorporated in the context of its local structural environment. Mapping such second-degree interactions is fundamentally difficult to do in physics-based models.
Based on this network approach, we developed a novel antibody-engineering platform to design and validate a pan anti-Dengue antibody that binds to and neutralizes all four serotypes of Dengue Viruses [8].

Figure 5. Residue contacts network
Comparison of a typical three-dimensional ribbon diagram of a representative glycan-binding protein (influenza A virus hemagglutinin) with its two-dimensional network diagram.